Введите слово или словосочетание на любом языке 👆
Язык:
Перевод и анализ слов искусственным интеллектом ChatGPT
На этой странице Вы можете получить подробный анализ слова или словосочетания, произведенный с помощью лучшей на сегодняшний день технологии искусственного интеллекта:
- как употребляется слово
- частота употребления
- используется оно чаще в устной или письменной речи
- варианты перевода слова
- примеры употребления (несколько фраз с переводом)
- этимология
Что (кто) такое Кинетика и катализ - определение
Кинетика химическая; Порядок реакции; Химкинетика; Кинетика химических реакций; Кинетика нулевого порядка; Кинетика первого порядка; Кинетика второго порядка



Найдено результатов: 6218
Кинетика и катализ
("Кине́тика и ката́лиз",)
научный журнал, орган Сибирского отделения АН СССР. Издается в Москве с 1960. Выходит 6 номеров в год. В журнале публикуются оригинальные теоретические и экспериментальные работы по кинетике химических превращений в газах, растворах и твердых фазах, по исследованию промежуточных активных частиц (радикалов, ионов), горению, механизму гомогенного и гетерогенного катализа, по научным основам подбора катализаторов, практически важным каталитическим процессам, влиянию процессов переноса вещества и тепла на кинетику химических превращении, по методике расчета и моделирования контактных аппаратов. Печатаются также обзоры по важнейшим вопросам катализа и кинетики химических превращений. Тираж (1972) 1650 экз.
ПОРЯДОК РЕАКЦИИ
по данному веществу , показатель степени при концентрации этого вещества в кинетическом уравнении химической реакции. Суммарный порядок реакции слагается из порядков реакции по всем веществам, концентрации которых входят в кинетическое уравнение.
Химическая кинетика
см. Кинетика химическая.
КИНЕТИКА ХИМИЧЕСКАЯ
раздел физической химии, учение о скоростях и механизмах химических реакций. Кинетика химическая - научная основа создания новых и совершенствования существующих процессов химической технологии. Методы кинетики химической используются в биологии и др. областях естествознания. См. также Макрокинетика.
Кинетика химическая
кинетика химических реакций, учение о химических процессах - о законах их протекания во времени, скоростях и механизмах. С исследованиями кинетики химических реакций связаны важнейшие направления современной химии и химической промышленности: разработка рациональных принципов управления химическими процессами; стимулирование полезных и торможение и подавление нежелательных химических реакций; создание новых и усовершенствование существующих процессов и аппаратов в химической технологии; изучение поведения химических продуктов, материалов и изделий из них в различных условиях применения и эксплуатации.
В реальных условиях, например в крупных промышленных аппаратах, химический процесс осложняется в связи с передачей тепла, выделяемого или поглощаемого в реакции, транспортом веществ в зону реакции, их искусственным или естественным перемешиванием. Эти проблемы решает так называемая Макрокинетика.
Вместе с тем многие уравнения, описывающие протекание во времени химических реакций, пригодны и для описания ряда физических процессов (распад радиоактивных ядер, деление ядерного горючего), а также для количественной характеристики развития некоторых биохимических, в том числе ферментативных, и других биологических процессов (нормальный и злокачественный рост тканей, развитие лучевого поражения, кинетические критерии оценки эффективности лечения). К. х. лежит в основе исследования сложных процессов горения газов и взрывчатых веществ, помогает изучению процессов в двигателе внутреннего сгорания. Таким образом, можно говорить об общей кинетике, частным случаем которой является кинетика химических реакций. Эти аналогии весьма удобны для практического использования, но всегда следует иметь в виду принципиальные различия в природе рассматриваемых явлений.
Ввиду сложности реальных химических систем и необходимости учета большого числа факторов и условий проведения процесса, при выяснении оптимальных режимов получения нужных продуктов в современной К. х. широко используются быстродействующие электронные вычислительные машины.
Историческая справка. Отдельные работы в области К. х. были выполнены ещё в середине 19 в. В 1850 немецкий химик Л. Вильгельми изучил скорость инверсии тростникового сахара, в 1862-63 М. Бертло - скорость реакций этерификации. В работах Н. А. Меншуткина получили развитие (1882-90) такие основные проблемы химии, как связь между строением веществ и их реакционной способностью, влияние среды на ход химического превращения. В 80-х гг. 19 в. Я. Вант-Гофф и С. Аррениус сформулировали основные законы, управляющие простыми химическими реакциями, и дали трактовку этих законов, исходя из молекулярно-кинетической теории. Дальнейшее развитие этих работ привело к созданию в 30-х гг. 20 в. Г. Эйрингом и М. Поляни на базе квантовой механики и статистической физики теории абсолютных скоростей реакций, открывающей перспективы расчёта скоростей простых (элементарных) реакций, исходя из свойств реагирующих частиц (см. Активированный комплекс).
Параллельно развивались работы по изучению кинетики сложных реакций. Среди первых в этой области были исследования А. Н. Баха и Н. А. Шилова по реакциям окисления. Они включили в предмет К. х. представления о решающей роли промежуточных продуктов и промежуточных реакций в химическом превращении. Большую роль в разработке общих методов подхода к изучению сложных реакций (См. Сложные реакции) сыграли работы М. Боденштейна. Выдающимся достижением теории сложных химических процессов явилась созданная в 30-х гг. Н. Н. Семеновым (См. Семёнов) общая теория цепных реакций (См. Цепные реакции). Широкие исследования механизма сложных кинетических процессов, особенно цепных реакций, были выполнены С. Н. Хиншелвудом.
Основные понятия и законы. Химическая реакция может протекать гомогенно, то есть в объеме одной фазы, и гетерогенно, то есть на границе раздела фаз. Наиболее полно разработана К. х. реакций в газовой фазе, так как она отправляется от хорошо развитой кинетической теории газового состояния. В то же время интенсивно развивается кинетика реакций в жидкой фазе и твердых телах. В зависимости от того, в какой форме подводится к реагирующей системе необходимая для реакций энергия (теплота, свет, электрический ток, излучение, плазма, лазерные пучки, высокие и сверхвысокие давления, ударные волны), они подразделяются на тепловые, фотохимические, электрохимические, радиационно-химические и др.
В основе К. х. как учения о скоростях химических превращений лежит Действующих масс закон, согласно которому скорость реакции веществ А, В, С,... пропорциональна произведению их концентраций. Скорость реакции характеризуется обычно изменением за единицу времени концентрации какого-либо из исходных веществ или конечных продуктов реакции. Например, скорость вступления в реакцию вещества А (уменьшение его концентрации в единицу времени) выражается уравнением:
- 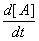 = k [A]α[B]β[C]γ...,
= k [A]α[B]β[C]γ...,
где к - константа скорости реакции, [А], [В], [С]... - концентрации реагирующих веществ (в качестве действующих веществ могут выступать молекулы, радикалы и ионы, в зависимости от типа реакции); знак минус показывает, что концентрация вещества А убывает со временем. Сумма величин α, β, γ... называется порядком реакции (См. Порядок реакции). В зависимости от числа молекул, участвующих в элементарном акте химического взаимодействия, различают реакции мономолекулярные, в которых реагируют отдельные молекулы одного вида, бимолекулярные - протекающие при двойном соударении (при встрече двух молекул), тримолекулярные - при тройном соударении. Реакции, требующие в элементарном акте встречи более трех молекул, мало вероятны. Порядок простой гомогенной реакции совпадает с числом молекул, участвующих в элементарном акте реакции. Однако чаще всего такого совпадения не бывает. В частности, показатели α, β, γ... могут быть дробными величинами. Это говорит о том, что реакция имеет сложный механизм, то есть протекает в несколько элементарных стадий, каждая из которых является строго моно-, би- или тримолекулярной реакцией. В тех случаях, когда сложная по существу реакция описывается простым кинетическим уравнением, говорят, что она имитирует простой закон протекания (см. Сложные реакции).
Температурная зависимость скорости реакции определяется уравнением Аррениуса: k-=k0e-E/RT,
где k0 - множитель, который в ряде простейших случаев может быть предвычислен, исходя из молекулярно-кинетических представлений о механизме элементарного акта, е - основание натуральных логарифмов, Е - Энергия активации реакции, R - универсальная газовая постоянная, Т - абсолютная температура.
На графически показано убывание со временем концентрации исходных веществ в случае реакций, удовлетворяющих простым законам. Кривые, показывающие изменение концентраций реагирующих веществ со временем, называются кинетическими кривыми.
По механизму химические процессы делятся на 3 основных типа: простые реакции между молекулами; радикальные, в том числе цепные реакции (протекающие через промежуточное образование свободных радикалов и атомов); ионные (идущие при участии ионов).
Кинетика реакций между молекулами. Реакции непосредственно между валентно-насыщенными молекулами весьма редки, т.к. происходящая при этом перестройка молекул требует разрыва химических связей, энергия которых достигает значительных величин (50-100 ккал/моль, или 209,3-418,7 кдж/моль). Поэтому в газовой фазе реакции идут чаще всего как цепные, а в жидкой фазе - и как цепные, и как ионные. Примерами реакций насыщенных молекул в газовой фазе могут служить: 1) мономолекулярная реакция (См. Мономолекулярные реакции) распада азометана: CH3N2CH3 → C2H6+N2; 2) бимолекулярная реакция (См. Бимолекулярные реакции) превращения йодистого нитрозила: NOI+NOI→2NO+I2 и 3) тримолекулярная реакция (См. Тримолекулярные реакции) окисления окиси азота в двуокись азота: 2NO+O2→2NO2.
Реакции, в которых превращение исходных веществ идёт по двум или нескольким направлениям, называются параллельными; механизм и кинетические закономерности реакций в разных направлениях могут быть самыми разнообразными - простыми и сложными (см. Параллельные реакции). Реакции, в которых превращение исходных веществ в конечные продукты происходит через несколько следующих друг за другом стадий с образованием промежуточных продуктов, называются последовательными (см. Последовательные реакции).
На показаны кинетические кривые для исходного, промежуточного и конечного веществ в последовательной реакции. Характерной особенностью этих кривых является наличие максимума у кривой промежуточного продукта и точки перегиба на кривой образования конечного продукта реакции. Однако эти особенности не могут служить однозначным признаком последовательной реакции. Известно много случаев, когда конечные продукты превращения ускоряют реакцию. Скорость таких автокаталитических процессов сначала возрастает вследствие увеличения количества продукта, являющегося катализатором, а затем уменьшается вследствие израсходования исходных веществ (см. Автокатализ). Реакция, идущая под влиянием другой, протекающей одновременно и в том же участке пространства, называется индуцированной, или сопряжённой (см. Сопряжённые реакции).
Кинетика цепных реакций. Реакции, в которых один первичный акт активации приводит к превращению большого числа молекул исходных веществ, называются цепными. В реакции зарождения цепи образуется активная частица - свободный радикал или атом. Эта активная частица реагирует с молекулой исходного вещества, образуя молекулу продукта реакции и (вследствие неуничтожимости свободной валентности) регенерируя новую активную частицу; образовавшийся радикал в свою очередь реагирует с исходной молекулой и т.д. (неразветвлённая цепь). Энергия активации взаимодействия радикалов и атомов с молекулами не превышает 10 ккал/моль (41,86 кдж/моль), поэтому длина цепи из элементарных химических реакций достигает тысяч и сотен тысяч звеньев. В некоторых цепных реакциях увеличивается число свободных валентностей, что приводит к появлению новых активных центров, то есть новых цепей. Таким образом, цепь разветвляется и реакция ускоряется (становится нестационарной).
Цепь обрывается в результате соединения (рекомбинации) двух радикалов, в случае реакции радикала с некоторыми примесными частицами, соударения со стенкой сосуда. Скорость неразветвленной цепной реакции вначале растет, затем достигает постоянного значения и, наконец, медленно убывает. Скорость разветвленной цепной реакции возрастает со временем и при благоприятных условиях может произойти воспламенение реагирующей смеси. Достигнув максимального значения, скорость реакции уменьшается из-за расходования исходных веществ (подробнее см. Цепные реакции). В соответствии с этим кинетические кривые цепных разветвленных процессов имеют характерную S-oбразную форму (). Точка перегиба на кривой отвечает максимуму скорости реакции.
Основы теории цепных реакций разработаны и экспериментально подтверждены в исследованиях советского ученого Н. Н. Семенова и его школы. В СССР успешно изучаются скорость и механизм важнейших групп цепных процессов: полимеризации, крекинга, окисления. На базе цепной теории окислительных реакций разработаны новые высокоэффективные технологические процессы получения важных химических продуктов (в частности, мономеров для получения полимеров) путем окисления нефтяного сырья и углеводородных газов. Цепная теория процессов ингибированного окисления позволяет предотвращать окислительную порчу (старение) полимеров, смазочных масел и бензинов, пищевых продуктов и лекарственных препаратов. Ингибиторы окисления, или стабилизаторы окислительных процессов (см. Ингибиторы химические), - это важнейшие представители малотоннажных продуктов органического синтеза.
Кинетика ионных реакций. Значительное число реакций в растворах протекает при участии ионов. Скорость ионных реакций сильно зависит от растворителей, так как в разных растворителях молекулы в разной степени диссоциированы на ионы. Энергия активации реакции ионов с молекулами невелика: заряд иона снижает энергию активации. При изучении кинетики реакций в растворах учитывают влияние полярных групп, наличие большого межмолекулярного взаимодействия, влияние растворителя и т.п.
Кинетика гетерогенных каталитических реакций. Для реакций газов и жидкостей, протекающих у поверхности твёрдых тел (см. Катализ), по-видимому, имеют место те же 3 основных типа химических превращений, которые были рассмотрены для гомогенных процессов, т. е. простые, радикально-цепные и ионные реакции. Различие заключается лишь в том, что в соответствующие кинетические уравнения входят концентрации реагирующих веществ в поверхностном адсорбционном слое (см. Адсорбция). Наблюдаются разные кинетические зависимости, которые обусловлены характером адсорбции исходных веществ и продуктов реакции на поверхности. Основной суммарный кинетический эффект катализатора заключается в снижении энергии активации реакции. Важной проблемой в области гетерогенного катализа является предвидение каталитического действия. Представления и методы, свойственные теории гетерогенного катализа, все больше сближаются с областью гомогенного катализа жидкофазных реакций, особенно при использовании в качестве катализаторов комплексных соединений переходных металлов. Выясняется механизм действия биологических катализаторов (ферментов), особенно с целью создания принципиально новых высокоэффективных катализаторов для химических реакций.
Советскими и зарубежными учёными успешно разрабатываются и многие другие актуальные проблемы К. х., например, применение квантовой механики к анализу элементарного акта реакции; установление связей между строением веществ и кинетическими параметрами, характеризующими их реакционную способность; изучение кинетики и механизма конкретных сложных химических реакций с применением новейших физических экспериментальных методов и современной вычислительной техники; использование кинетических констант в инженерных расчётах в химической и нефтехимической промышленности.
Лит.: Семенов Н. Н., О некоторых проблемах химической кинетики и реакционной способности, 2 изд., М., 1958; Кондратьев В. Н., Кинетика химических газовых реакций, М., 1958; Эмануэль Н. М., Кнорре Д. Г., Курс химической кинетики, 2 изд., М., 1969; Бенсон С., Основы химической кинетики, пер. С англ., М., 1964: Эмануэль Н. М., Химическая кинетика, в сборнике: Развитие физической химии в СССР, М., 1967.
Н. М. Эмануэль.
Порядок реакции
понятие кинетики химической (См. Кинетика химическая). П. р. определяется как сумма показателей степеней n1 и n2 в уравнении
выражающем зависимость скорости реакции r от концентраций [A1] и [А2] исходных веществ (k - константа скорости). Реакции с n1 + n2 = 1, 2 и т.д. называются реакциями 1-го, 2-го и т.д. порядков. Отдельный показатель степени в уравнении (1) называется порядком реакции по соответствующему веществу.
У простых реакций скорость в одном направлении, согласно Действующих масс закону, подчиняется уравнению (1), а n1 и n2 совпадают с числом молекул веществ A1 и А2, участвующих в элементарном акте реакции. Скорости сложных реакций (См. Сложные реакции) иногда также выражаются уравнениями вида (1), но при этом П. р. может не совпадать со стехиометрическим коэффициентом вещества в уравнении реакции (записанном с наименьшими целочисленными стехиометрическими коэффициентами), может быть дробным числом. В гетерогенно-каталитических реакциях обычны наряду с целыми дробные и нулевые порядки; встречаются также отрицательные порядки.
М. И. Тёмкин.
Химическая кинетика
Химическая кинетика или кинетика химических реакций — раздел физической химии, изучающий закономерности протекания химических реакций во времени, зависимости этих закономерностей от внешних условий, а также механизмы химических превращений.
КИНЕТИКА ФИЗИЧЕСКАЯ
МИКРОСКОПИЧЕСКАЯ ТЕОРИЯ ПРОЦЕССОВ В НЕРАВНОВЕСНЫХ СРЕДАХ
Кинетическая теория газов; Кинетика физическая
раздел статистической физики, в котором изучаются на основе молекулярно-кинетической теории неравновесные процессы в веществе, напр. процессы выравнивания концентраций в смесях (диффузия), температур (теплопроводность) и т. д.
Кинетика физическая
МИКРОСКОПИЧЕСКАЯ ТЕОРИЯ ПРОЦЕССОВ В НЕРАВНОВЕСНЫХ СРЕДАХ
Кинетическая теория газов; Кинетика физическая
теория неравновесных макроскопических процессов, то есть процессов, возникающих в системах, выведенных из состояния теплового (термодинамического) равновесия. К К. ф. можно отнести термодинамику неравновесных процессов (См. Термодинамика неравновесных процессов), кинетическую теорию газов (См. Кинетическая теория газов) (в том числе плазмы), теорию процессов переноса в твёрдых телах, а также общую статистическую теорию неравновесных процессов, которая начала развиваться лишь в 50-е гг.
Все неравновесные процессы в адиабатически изолированных системах (системах, не обменивающихся теплом с окружающими телами) являются необратимыми процессами (См. Необратимые процессы) - происходят с увеличением энтропии (См. Энтропия); в равновесном состоянии энтропия достигает максимума.
Как и в случае равновесных состояний, в К. ф. возможны два способа описания систем: феноменологический, или термодинамический (термодинамика неравновесных процессов), и статистический.
Термодинамический метод описания неравновесных процессов
При термодинамическом описании неравновесных процессов рассматривается изменение в пространстве и времени таких макроскопических параметров состояния системы, как плотность массы i-го компонента ρi (r, t), плотность импульса ρu (r, t), локальная температура T (r, t), поток массы i-го компонента ji (r, t), плотность потока внутренней энергии q (r, t) [здесь r - координата, t - время, u - средняя массовая скорость, ρ - плотность массы]. В равновесном состоянии системы ρ, ρi, Т постоянны, а потоки равны нулю.
Термодинамическое описание неравновесных возможно лишь при достаточно медленном параметров состояния в пространстве и во времени для состояний, близких к равновесным. Для газов это означает, что все термодинамические параметры, характеризующие состояние системы, мало меняются на длине свободного пробега и за время, равное среднему времени свободного пробега молекул (среднему времени между двумя последовательными столкновениями молекул). Медленные процессы встречаются практически очень часто, так как установление равновесия происходит только после очень большого числа столкновений; к ним относятся: Диффузия, Теплопроводность, Электропроводность и т.д. Отклонения от состояния термодинамического равновесия характеризуются Градиентами температуры, концентрации (ρi/ρ) и массовой скорости (так называемыми термодинамическими силами), а потоки энергии, массы i-го компонента и импульса связаны с термодинамическими силами линейными соотношениями. Коэффициенты в этих соотношениях называются кинетическими коэффициентами.
Рассмотрим в качестве примера диффузию в бинарной смеси, то есть процесс выравнивания концентрации компонентов в результате хаотического теплового движения молекул. Феноменологическое уравнение, описывающее процесс диффузии, получают с помощью закона сохранения вещества и того опытного факта, что поток вещества одного из компонентов вследствие диффузии прямо пропорционален градиенту его концентрации (с обратным знаком). Коэффициент пропорциональности называется коэффициентом диффузии. Согласно уравнению диффузии, скорость изменения концентрации вещества со временем прямо пропорциональна дивергенции (См. Дивергенция) градиента концентрации с коэффициентом пропорциональности, равным коэффициенту диффузии.
Решение уравнения диффузии позволяет определить время, в течение которого произойдёт выравнивание концентрации молекул в системе (например, в сосуде с газом) за счёт диффузии (время релаксации). Время релаксации τр имеет порядок: τр Кинетика физическая L2/D, где L - линейные размеры сосуда, a D - коэффициент диффузии. Это время тем больше, чем больше размеры сосуда и чем меньше коэффициент диффузии. Коэффициент диффузии пропорционален длине свободного пробега молекул λ и их средней тепловой скорости ν. Поэтому время релаксации оказывается пропорциональным: τр Кинетика физическая L2/ λν = (L/λ)2λ/ν, где λ/ν = τ - среднее время свободного пробега. Очевидно, что τр >> τ при L >> λ. Таким образом, условие L >> λ (размеры системы велики по сравнению с длиной свободного пробега молекул) является необходимым для того, чтобы процесс установления равновесного состояния можно было считать медленным. Аналогичным образом устанавливаются уравнения, описывающие теплопроводность, внутреннее трение, электропроводность и т.д. Коэффициент диффузии, теплопроводности и вязкости, а также удельная электропроводность в феноменологической теории должны быть определены экспериментально.
Перечисленные процессы называются прямыми. Этим подчёркивается, что, например, при диффузии градиент концентрации данного вещества вызывает поток этого же вещества; градиент температуры вызывает поток внутренней энергии, которая при постоянной концентрации молекул меняется только с температурой; электрический ток вызывается градиентом потенциала и т.д. Кроме прямых процессов, существуют ещё так называемые перекрёстные процессы. Примером перекрёстного процесса может служить Термодиффузия - перенос вещества не вследствие градиента концентрации (это была бы обычная диффузия), а вследствие градиента температуры. Термодиффузия создаёт градиент концентрации, что приводит к появлению обычной диффузии. Если разность температур в системе поддерживается постоянной, то устанавливается стационарное состояние, при котором потоки вещества, вызванные градиентами температуры и концентрации, взаимно уравновешиваются. В смеси газов при этом концентрация молекул в местах повышенной температуры оказывается большей для молекул меньшей массы (данное явление используется для разделения изотопов (См. Изотопы)).
Градиент концентрации в свою очередь создаёт поток внутренней энергии. В этом состоит процесс диффузионной теплопроводности. При наличии в теле заряженных частиц градиент температуры создаёт упорядоченное перемещение этих частиц - электрический ток, называемый термоэлектрическим (см. Термоэлектрические явления).
В К. ф. важное значение имеет принцип симметрии кинетических коэффициентов, установленный Л. Онсагером. В равновесном состоянии термодинамические параметры ai (давление, температура и т.д.), характеризующие состояние макроскопической системы, постоянны во времени: dai/dt = 0. Важнейшая функция состояния системы - энтропия S, зависящая от ai, в состоянии равновесия имеет максимум и, следовательно, её частные производные ∂S/∂aj = 0. При малом отклонении системы от равновесия производные ∂S/∂aj и ∂a/∂t малы, но отличны от нуля, и между ними существуют приближённые линейные соотношения. Коэффициенты пропорциональности в этих соотношениях и есть кинетические коэффициенты. Если через γik обозначить коэффициент, определяющий скорость изменения параметра системы ai зависимости от 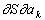 , то, согласно принципу Онсагера (в отсутствие магнитного поля и вращения системы как целого), имеет место равенство γ ik = γ ki. Принцип Онсагера вытекает из свойства микроскопической обратимости, которая выражается в инвариантности уравнений движения частиц системы относительно замены знака времени: t → -t (см. Онсагера теорема). Из этого принципа, в частности, следует существование связи между коэффициентами, определяющим выделение током тепла из-за неравномерного нагрева проводника (Томсона эффект), и коэффициентами, определяющим выделение током тепла в спаях разнородных проводников или полупроводников (Пельтье эффект).
, то, согласно принципу Онсагера (в отсутствие магнитного поля и вращения системы как целого), имеет место равенство γ ik = γ ki. Принцип Онсагера вытекает из свойства микроскопической обратимости, которая выражается в инвариантности уравнений движения частиц системы относительно замены знака времени: t → -t (см. Онсагера теорема). Из этого принципа, в частности, следует существование связи между коэффициентами, определяющим выделение током тепла из-за неравномерного нагрева проводника (Томсона эффект), и коэффициентами, определяющим выделение током тепла в спаях разнородных проводников или полупроводников (Пельтье эффект).
Статистический метод описания неравновесных процессов.
Статистическая теория неравновесных процессов является более детальной и глубокой, чем термодинамическая. В отличие от термодинамического метода, статистическая теория на основе определенных представлений о строении вещества и действующих между молекулами силах позволяет вычислить кинетические коэффициенты, определяющие интенсивность процессов диффузии, внутреннего трения (вязкости (См. Вязкость)), электропроводности и т.д. Однако эта теория весьма сложна.
Статистический метод описания систем как в равновесном, так и неравновесном состоянии основан на вычислении функции распределения. Для равновесных состояний имеются универсальные функции распределения координат и импульсов (или скоростей) всех частиц, определяющие вероятность того, что эти величины принимают фиксированные значения. Для систем, находящихся в тепловом контакте с окружающей средой, температура которой постоянна, это - каноническое Гиббса распределение, а для изолированных систем - микроканоническое Гиббса распределение; оба распределения полностью определяются энергией системы.
Неравновесные состояния в гораздо большей степени (чем равновесные) зависят от микроскопических свойств систем: свойств атомов и молекул и сил взаимодействия между ними. Лишь в 50-60-е гг. были разработаны общие методы построения функций распределения (по координатам и импульсам всех частиц системы), аналогичных каноническому распределению Гиббса, но описывающих неравновесные процессы.
С помощью функций распределения можно определить любые макроскопические величины, характеризующие состояние системы, и проследить за их изменением в пространстве с течением времени. Это достигается вычислением статистических средних (см. Статистическая физика). Нахождение функции распределения, зависящей от координат и импульсов всех частиц, является в общем случае неразрешимой задачей, т.к. оно эквивалентно решению уравнений движения для всех частиц системы. Однако для практических целей нет необходимости в знании точного вида этой функции распределения: она содержит слишком подробную информацию о движении отдельных частиц, которая не существенна для определения поведения системы в целом. В связи с этим используется приближенное статистическое описание с помощью более простых функций распределения. Для описания состояния газов средней плотности достаточно знания так называемой одночастичной функции распределения f (p, r, t), дающей среднее число частиц с определёнными значениями импульсов р (или скоростей ν) и координат r. Для газов более высокой плотности необходимо знание двухчастичных (парных) функций распределения. Общий метод получения уравнений для одночастичных и более сложных функций (зависящих от координат и импульсов двух и более частиц) был разработан Н. Н. Боголюбовым, М. Борном, М. Грином и др. Эти уравнения называются кинетическими. К их числу относится Кинетическое уравнение Больцмана для разреженных газов, полученное Л. Больцманом из соображений, основанных на балансе частиц со скоростями в интервалах Δνx, Δνy, Δνz внутри объёма Δх Δy Δz (νx, νy, νz - проекции скорости ν на координатные оси х, у, z). Разновидностями уравнения Больцмана для ионизированного газа (плазмы) являются кинетические уравнения Л. Д. Ландау и А. А. Власова (см. Плазма).
Кинетические уравнения могут быть построены не только для газов, но и для малых возбуждений в конденсированных системах. Тепловое движение системы характеризуется различного рода возбуждениями. В газе это - поступательное движение составляющих его частиц и внутренние возбуждения атомов и молекул. В общем случае тепловое движение характеризуется возбуждениями более сложной природы. Так, в кристаллических телах тепловое возбуждение можно представить в виде упругих волн, распространяющихся вдоль кристалла, точнее - волн, соответствующих нормальным колебаниям кристаллической решётки (См. Колебания кристаллической решётки). В плазме коллективными возбуждениями являются колебания плотности электрического заряда, вызванные дальнодействующими кулоновскими силами. В металлах возможны электронные возбуждения (переходы электронов из состояний внутри Ферми поверхности (См. Ферми поверхность) в состояния вне её), а в полупроводниках - ещё и дырочные возбуждения (появление свободных от электронов состояний в валентной зоне при переходе электронов в зону проводимости; см. Полупроводники). При низких температурах, в слабовозбуждённом состоянии, энергию возбуждения всегда можно представить в виде суммы некоторых элементарных возбуждений, или, на квантовом языке, квазичастиц (См. Квазичастицы). Понятие о квазичастицах применимо не только для кристаллических тел, но и для жидких, газообразных и аморфных, если температура не слишком велика. Функции распределения для квазичастиц системы, находящейся в неравновесном состоянии, удовлетворяют кинетическому уравнению.
В случае квантовых систем функция распределения зависит от Спина частиц (или квазичастиц). В частности, для частиц с полуцелым спином равновесной функцией распределения служит распределение Ферми - Дирака, а для частиц (квазичастиц) с целым или нулевым спином - распределение Бозе - Эйнштейна (см. Статистическая физика).
В кинетических уравнениях наряду с внешними воздействиями учитываются взаимодействия между частицами или квазичастицами, причем эти взаимодействия рассматриваются как парные столкновения. Именно эти взаимодействия приводят к установлению равновесных состояний. Во многих случаях функция распределения не зависит явно от времени. Такая функция называется стационарной, она описывает процессы, течение которых не претерпевает изменений со временем. При стационарных процессах изменение функции распределения вследствие внешних воздействий компенсируется её изменением в результате столкновений.
В простых случаях можно грубо оценить изменение функции распределения f системы в результате столкновений, считая, что оно пропорционально величине отклонения от равновесной функции (так как только при отклонении от состояния равновесия столкновения меняют функцию распределения). Величина, обратная коэффициенту пропорциональности в этом соотношении, называется временем релаксации. В общем случае учесть взаимодействие таким простым способом невозможно, и в кинетическое уравнение входит так называемый интеграл столкновений, который более точно учитывает результат изменения функции распределения вследствие взаимодействия частиц (квазичастиц).
Решая кинетическое уравнение, находят неравновесную функцию распределения и вычисляют потоки энергии, массы и импульса, что позволяет получить уравнения теплопроводности, диффузии и переноса импульса (уравнение Навье - Стокса) с кинетическими коэффициентами, выраженными через молекулярные постоянные. [Однако кинетическое уравнение можно построить лишь для газов (из частиц или квазичастиц)].
Основные принципы теории неравновесных процессов надёжно установлены. Разработаны методы построения уравнений переноса энергии, массы и импульса в различных системах, не только в газах, а, например, и в жидкостях. При этом получают выражения для кинетических коэффициентов, входящих в эти уравнения, через корреляционные функции (функции, описывающие корреляцию в пространстве и во времени) потоков этих физических величин, то есть в конечном счете, через молекулярные постоянные. Эти выражения очень сложны и могут быть вычислены лишь средствами современной вычислительной математики.
Лит.: Гуревич Л. Э., Основы физической кинетики, М.- Л., 1940; Боголюбов Н. Н., Проблемы динамической теории в статистической физике, М.-Л., 1946; Гуров К. П,, Основания кинетической теории. Метод Н. Н. Боголюбова, М., 1966; Ландау Л. Д., Лифшиц Е. М., Статистическая физика, М., 1964 (Теоретическая физика, т. 5): Климонтович Ю. Л., Статистическая теория неравновесных процессов в плазме, М., 1964; Пригожин И. Р., Неравновесная статистическая механика, пер. с англ., М., 1964; Зубарев Д. Н., Неравновесная статистическая термодинамика, М., 1971; Гроот С., Мазур П., Неравновесная термодинамика, пер. с англ., М., 1964; Честер Дж., Теория необратимых процессов, пер. с англ., М., 1966; Хаазе Р., Термодинамика необратимых процессов, пер. с нем., М., 1967.
Г. Я. Мякишев.
Рис. 1. Кинетические кривые химических реакций простых типов.
Рис. 2. Изменение концентрации исходного 1, промежуточного 2 и конечного 3 веществ в последовательной реакции.
Рис. 3. Типичная кинетическая кривая цепного разветвленного процесса. Формально аналогичный вид имеют и кривые автокаталитических реакций.
Физическая кинетика
МИКРОСКОПИЧЕСКАЯ ТЕОРИЯ ПРОЦЕССОВ В НЕРАВНОВЕСНЫХ СРЕДАХ
Кинетическая теория газов; Кинетика физическая
Физи́ческая кине́тика ( — движение) — микроскопическая теория процессов в неравновесных средах. В кинетике методами квантовой или классической статистической физики изучают процессы переноса энергии, импульса, заряда и вещества в различных физических системах (газах, плазме, жидкостях, твёрдых телах) и влияние на них внешних полей.
Википедия
Химическая кинетика
Химическая кинетика или кинетика химических реакций — раздел физической химии, изучающий закономерности протекания химических реакций во времени, зависимости этих закономерностей от внешних условий, а также механизмы химических превращений.
Предметом химической кинетики является изучение всех факторов, влияющих на скорость как суммарного процесса, так и всех промежуточных стадий.

